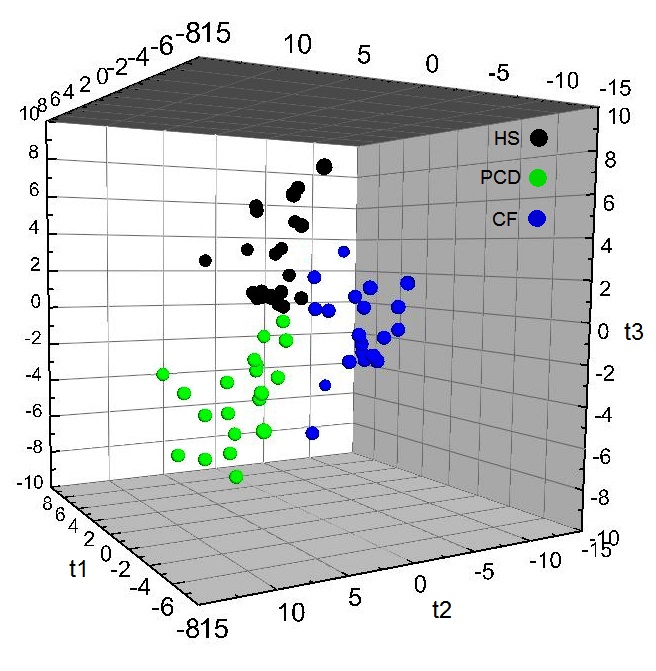
Un’analisi non invasiva permetterà di differenziare la discinesia ciliare e la fibrosi cistica, le più diffuse malattie croniche a carico dell’apparato respiratorio, che presentano sintomi simili e sono progressive e invalidanti. Lo studio, realizzato dai ricercatori dell’Icb-Cnr, è pubblicato sull’American Journal of Respiratory and Critical Care Medicine. Sarà finalmente possibile differenziare mediante un’analisi non invasiva, la spettroscopia di risonanza magnetica (Nmr), due malattie genetiche che presentano sintomatologie simili: la discinesia ciliare e la fibrosi cistica. Il risultato è stato ottenuto grazie a uno studio di un gruppo dell’Istituto di chimica biomolecolare del Consiglio nazionale delle ricerche (Icb-Cnr) di Pozzuoli (Na). Il lavoro è pubblicato su ‘American Journal of Respiratory and Critical Care Medicine’. “Discinesia ciliare (Dcp) e fibrosi cistica (Fc) sono due malattie genetiche che colpiscono l’apparato respiratorio e provocano la degenerazione di altri organi, manifestando sintomi clinico-patologici sovrapponibili”, spiega Andrea Motta dell’Icb-Cnr, che ha diretto il gruppo. “Il nostro studio ha indicato la possibilità di ottenere, tramite il condensato dell’aria espirata, un tracciato metabolico polmonare. Combinando la spettroscopia di risonanza magnetica nucleare con le tecniche di analisi statistica, è possibile costruire dei modelli matematici affidabili che ci permettono di caratterizzare le alterazioni metaboliche specifiche di ciascuna patologia”. Le due malattie croniche sono in genere progressive e altamente invalidanti. “Nella Fc, che è la malattia genetica ereditaria mortale più comune nella popolazione caucasica, l’alterazione nell’escrezione del cloro porta alla secrezione di muco molto denso e viscoso e quindi poco scorrevole, con notevoli infezioni polmonari e con il coinvolgimento di differenti organi interni”, prosegue il ricercatore. “La Dcp, invece, è caratterizzata da alterazioni della struttura e/o della funzione delle ciglia della mucosa respiratoria con conseguente difetto del trasporto muco-ciliare ed è, per frequenza, la seconda malattia congenita dell’apparato respiratorio dopo la fibrosi cistica”. Lo studio, cofinanziato dalla Fondazione per la ricerca sulla fibrosi cistica, vede la partecipazione dell’Università Federico II di Napoli (Francesca Santamaria), della Cattolica di Roma (Paolo Montuschi), dell’ospedale pediatrico Bambin Gesù di Roma (Vincenzina Lucidi) e dell’Imperial College di Londra (Andrew Bush). “Il nostro approccio è estremamente sensibile ed è in grado di valutare differenze metaboliche in patologie con stati fisiopatologici sovrapponibili. La possibilità di poter conoscere, tramite condensato espirato, la loro evoluzione, apre la strada a nuove strategie terapeutiche basate su un approccio personalizzato in cui il paziente segue una terapia impostata esclusivamente sulle sue necessità mediante trattamenti farmacologici mirati”, conclude Motta.
Ultime News
Magazine Online di Informazione ed Eventi